Open Access is an initiative that aims to make scientific research freely available to all. To date our community has made over 100 million downloads. It’s based on principles of collaboration, unobstructed discovery, and, most importantly, scientific progression. As PhD students, we found it difficult to access the research we needed, so we decided to create a new Open Access publisher that levels the playing field for scientists across the world. How? By making research easy to access, and puts the academic needs of the researchers before the business interests of publishers.
We are a community of more than 103,000 authors and editors from 3,291 institutions spanning 160 countries, including Nobel Prize winners and some of the world’s most-cited researchers. Publishing on IntechOpen allows authors to earn citations and find new collaborators, meaning more people see your work not only from your own field of study, but from other related fields too.
Molecular docking software is an indispensable tool in the dynamic landscape of computational biology and drug discovery. This software facilitates the simulation and prediction of interactions between small molecules and target proteins, offering a detailed glimpse into molecular binding phenomena. Employing sophisticated algorithms, these programs assess binding affinity, predict binding modes, and contribute crucial insights to streamline drug development processes. Examples of widely used molecular docking software include AutoDock, GOLD, and DOCK, each renowned for their capabilities in optimizing lead compounds, virtual screening, and elucidating the intricate details of protein-ligand interactions. Researchers leverage these tools to expedite the identification of potential drug candidates, ultimately bridging the gap between theoretical predictions and experimental advancements in the pursuit of novel therapeutics.
Faculty of Pharmacy, Integral University, Lucknow, Uttar Pradesh, India
Mohammad Ahmad
Faculty of Pharmacy, Integral University, Lucknow, Uttar Pradesh, India
Sahil Hussain
Faculty of Pharmacy, Integral University, Lucknow, Uttar Pradesh, India
Mohemmed Faraz Khan*
Faculty of Pharmacy, Integral University, Lucknow, Uttar Pradesh, India
*Address all correspondence to: faraz91khan@gmail.com
1. Introduction
Amidst the binary symphony of code and the precision of algorithms, Molecular Docking Software emerges as a crucial instrument in the scientific toolkit, within the digital confines of computational biology. The landscape of molecular docking has evolved over the past thirty years, spurred by the demands of structural molecular biology and the pursuit of structure-based drug discovery. This evolution finds its roots in the remarkable expansion of computational capabilities, coupled with the increasing accessibility of databases housing small molecules and proteins. The synergy of these advancements has propelled the field forward, opening avenues for deeper exploration and understanding [1, 2, 3]. Automated molecular docking software sets out to unravel and foresee molecular recognition on two fronts: structurally, by identifying probable binding modes, and energetically, by forecasting binding affinity. This computational endeavor predominantly occurs in the dynamic interplay between a small molecule and a target macromolecule [4]. The objective is to decode the intricate language of molecular interactions, providing insights into the binding intricacies that govern biochemical processes. Frequently termed as ligand-protein docking, there is an increasing fascination with the exploration of protein–protein docking. The applications of molecular docking in drug discovery are diverse and encompass a spectrum of functions. These include conducting structure–activity studies, optimizing lead compounds, identifying potential leads through virtual screening, offering binding hypotheses to support predictions for mutagenesis studies, aiding x-ray crystallography by fitting substrates and inhibitors to electron density, facilitating chemical mechanism studies, and contributing to the design of combinatorial libraries. The versatility of molecular docking underscores its pivotal role across various stages of the drug development process [5, 6]. Simulations reveal biological disorder impacts (mutation effects, phosphorylation, protonation states) in various complexes: protein–protein, protein-ligand, protein-peptide, or protein-nucleic acid interactions [7]. In this section, our emphasis has been on molecular docking, the underlying algorithms, and the execution of simulations. Utilizing virtual screening grounded in molecular descriptors and physicochemical properties of both active and inactive ligands proves highly valuable in identifying hits and leads through library enrichment for screening [8]. This approach is also effective in streamlining and enhancing ligand libraries for molecular docking, with emerging evidence suggesting that ligand shape matching can be as effective as, if not more effective than, docking. Nonetheless, when molecular docking serves as the concluding phase in virtual screening, it contributes essential 3D (three-dimensional) structural hypotheses elucidating how a ligand engages with its target [9].
Molecular docking uses a variety of SF (scoring functions), it is an in-silico technique that identifies the proper binding pose of a protein-ligand complex and assesses its strength to choose the optimal posture produced by each molecule to a rank order [10]. Docking techniques strive to seamlessly position a ligand within the binding site of a target protein. This involves the harmonization and optimization of variables such as hydrophobic, steric, and electrostatic complementarity, thereby providing an estimate of their binding free energy [11]. Molecular docking encapsulates three primary objectives: virtual screening, pose prediction, and the estimation of binding affinity [12]. A dependable docking technique should effectively distinguish between binding and non-binding sites, discerning their molecular interactions. Moreover, when handling extensive compound libraries, the method must aptly differentiate binding molecules from non-binding ones and accurately rank the binding molecules amongst the top compounds in the database [13]. The efficacy of virtual screening hinges on the quantity and precision of structural information available for the target protein and the ligand undergoing docking. The initial phase involves scrutinizing the target to identify the presence of pertinent binding sites [14]. This analysis is accomplished by studying established protein-ligand co-crystal structures or employing in-silico methods to pinpoint novel binding pockets [15]. Similar pockets are found using techniques such as the catalytic site AFT [16], SURFACE [17], POCKET SURFER, PATCH-SURFER, and Atlas. A “spherical probe,” or a pre-computer depiction of an idealized ligand, is rolled over the grid surface of another in-silico approach called POCKET to identify the binding site of a target protein. Moreover, various small molecule databases, including ZINC, PubChem, ChemDB, and DrugBank, collectively host over a million deposited structures of potential ligands. Docking has become an essential virtual screening process, particularly when thousands or even millions of compounds need to be evaluated against one or more targets in a short amount of time from a database. Replicating such a search would be unfeasible in terms of time and money experimentally. In docking simulations, achieving a successful outcome relies on the pivotal features of speed and accuracy. The primary goal in developing a docking algorithm is to create a swift method capable of identifying novel lead compounds during virtual screening or accurately reproducing experimental conformations for validation with experimental data. Numerous docking programs include ZDOCK [18], M-ZDOCK [19], MS-DOCK [20], Surflex [21], MCDOCK [22], GOLD, AUTODOCK [23], and others.
Every docking program uses a different search technique, such as IC (Incremental Construction) [24], GA (Genetic Algorithm) [25], MC (Monte Carlo) [26], and others. As explained in more detail in the section that follows, each has a distinct parameter set and search strategy. The receptor and ligand input coordinates are used as a set of parameters by the docking algorithm, which searches for the best possible match between two or more molecules. These factors include the flexibility of receptor or ligand structures, as well as geometric complementarity that takes into consideration the charge and radius of atoms in the VDW (van der Waals). Interatomic interactions are also taken into account, including hydrophobic contacts and hydrogen bonds. The expected alignments (poses) of a ligand within the target’s binding site are the result of docking applications. The posing usually results in several reasonable conformations. SFs that can assess binding free energy or intermolecular binding affinity are used to improve and rank these results. This optimization process culminates in the selection of the best orientation after the docking procedure.
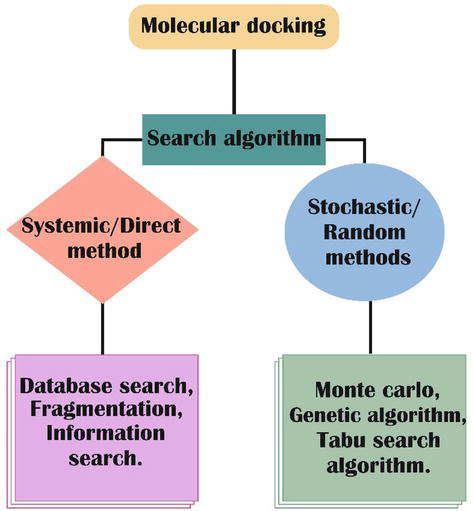
Various molecular docking programs are constructed based on a range of algorithms, each offering unique advantages. This overview focuses on highlighting the most prominent algorithms and their associated benefits. Additionally, we provide insights into key docking programs developed around these algorithms. It is worth noting that these programs often leverage one or more specific search algorithms in their operations. To facilitate understanding, the categorization of these search algorithms, as depicted in Figure 1, serves as a structured framework for comprehending their functionalities and applications:
A systematic or direct approach: The following are the three subtypes of systematic methods:
Conformational search: The structural property of the ligand is gradually adjusted in its torsional (dihedral), translational, and rotational degrees of freedom.
Fragmentation: In this case, the fragments can be anchored individually, with the first fragment being docked initially and the remaining fragments being constructed outward in stages from that initial bound point, or they can be docked together to create connections during the molecular docking process. It employs tools such as DOCK, LUDI, Flex XTM, etc.
Database Search: With this method, all the small molecules that are already registered in the database may be created in a large number of appropriate conformations, which can then be docked as hard bodies. One of the instruments it uses is FLOG.
Stochastic methods or Random methods: Three subcategories of stochastic approaches exist.
Monte Carlo: This method entails scoring the receptor binding site by randomly inserting ligands, after which a new configuration is produced. It uses instruments like ICM, MCDOCK, and so on.
Genetic algorithm: The “gene” describes the configuration and placement of the receptor concerning a population of postures, whilst the “fitness” determines the score. To create the following generation and repeat the agreement, carry out transformations, hybrids, etc. of the fittest. It employs applications like AutoDock, GOLD, and others.
Tabu search: It works by imposing restrictions that make it easier to study a new configuration by avoiding further examination of the ligands’ conformational space’s previously revealed regions. It makes use of MVD (Molegro Virtual Docker) TM, PRO LEADS, and other technologies [27].
Figure 1.
Classification of the search algorithm in molecular docking.
3.1 Fast shape matching
Fast shape matching algorithms consider the geometric overlap between two molecules, employing various methods to generate alignments between ligand and receptor. This approach can identify potential binding sites of a protein through a macromolecular surface search. One prominent fast shape matching algorithm used in molecular docking is the Shape Complementarity principle, which evaluates the geometric fit between the ligand and the protein’s binding site.
The application of fast shape matching algorithms significantly accelerates the initial screening process in molecular docking studies, enabling the rapid identification of potential ligands that fit well into the target protein’s binding site. By efficiently filtering out poorly fitting ligands, these algorithms help prioritize promising candidates for further optimization and experimental validation. Additionally, they play a crucial role in virtual screening campaigns aimed at identifying lead compounds for drug discovery. Moreover, these algorithms establish plausible conformations of predicted binding sites [28]. For instance, the initial steps of the DOCK program involve delineating the regions of the binding site where a potential ligand could be situated. This is achieved by identifying sphere centres through a Matching algorithm. Notably, this approach is also utilized in many other widely recognized programs, including DOCK itself [29].
3.2 Monte Carlo algorithm
MC algorithms are stochastic computational methods used for sampling from probability distributions to solve various types of problems, including optimization and integration. In molecular docking, it is often employed to explore the conformational space of ligands and find energetically favorable binding poses with target proteins.
MC simulations were initially introduced as a minimization process in molecular dynamics applications, exemplified by implementations in GROMACS and GROMOS [30, 31]. Subsequently, they found adaptations for flexible docking algorithms like MCDOCK and ICM. Additionally, MC simulations have demonstrated efficacy in accurately determining relative binding constants for protein systems [32]. This method is great for studying different thermodynamic conditions in biomolecular systems, but it is not good for analyzing time-dependent processes like molecular dynamics simulations. But with flexible docking programs, the search algorithm tries out various orientations and rotations to dock the ligand into the receptor site, reducing the chance of getting stuck in local minima. Stochastic techniques are used to determine potential ligand orientations inside the binding site, generating semi-random number sequences in MC simulations. If needed, these sequence generators can recreate the sequence and make sets of independent integers.
The MC methods rely on the Metropolis algorithm, which furnishes an acceptance criterion in the evolution of the docking search. At each iteration of the algorithm, a random variation of the ligand’s degrees of freedom is introduced [33]. The change is accepted when the energy score of the binding poses increases; otherwise, it is accepted based on the P (probability) expressed in the following equation.
Paccept=min(1,exp(−kTΔE))
Where, P accept is the probability of accepting a proposed conformation. ΔE is the change in energy associated with the proposed conformation. k is the Boltzmann constant. T is the temperature of the system [34].
3.3 Particle swarm optimization
Molecular docking employs swarm optimization techniques as a metaheuristic, serving as an effective search strategy for addressing the intricacies of the docking problem. Here, the docking problem is framed as an optimization task, where various parameters are associated with a well-defined SF. The primary objective is to ascertain the optimal conformation of a ligand within the receptor site, with the aim of minimizing energy interactions.
PSO (Particle Swarm Optimization) has emerged as a rapid and precise method uniquely suited to tackle the complexities inherent in molecular docking. This approach facilitates the exploration of the vast conformational space effectively, seeking the most favorable ligand-receptor interaction configurations. Numerous docking programs, such as SODOCK, are developed based on swarm principles, harnessing the collective intelligence of a swarm of particles to navigate the search space efficiently.
The PSO principle aligns seamlessly with the requirements of molecular docking, particularly emphasizing the minimization of the ligand’s energy to optimize its binding affinity according to the specified SF. This synergy between PSO and molecular docking enhances the efficiency and accuracy of the docking process, making it a preferred choice in computational drug discovery and protein-ligand interaction studies.
3.4 Genetic algorithm
A GA is a search heuristic inspired by the principles of natural selection and genetics. It is commonly used to find approximate solutions to optimization and search problems. Molecular docking, on the other hand, is a computational technique used in structural biology and bioinformatics to predict how two or more molecules interact with each other.
The application of GA in molecular docking involves using the principles of natural selection to optimize the orientation and conformation of ligands within the binding site of a target protein. The goal is to find the best possible binding pose, which corresponds to the configuration where the ligand binds most tightly to the protein [35].
GA proves valuable in identifying the best parameters influencing the activity of the studied drug molecule and enhancing computational efficiency. Inspired by Darwin’s theory of natural evolution, GA employs a genetic operator to combine two chromosomes (parents) and generate a new chromosome that may outperform its parents [36, 37]. This process incorporates multiple SF along with a set of parameters such as crossover rate and mutation rate. So there are many software applications that employ GA and one of them is GOLD (Genetic Optimization for Ligand Docking), which is a molecular docking software that employs a GA to explore the conformational space of ligands and predict their binding modes with a target protein.
Molecular docking tools are computational programs designed to predict the binding mode and affinity of a ligand to its target protein. These tools employ various algorithms to explore the spatial and energetic complementarity between the ligand and the protein binding site. By simulating the docking process, they assist researchers in understanding the molecular interactions underlying biological processes and aid in drug discovery efforts. Table 1 provides an overview of some well-known docking tools along with the algorithms they utilize. These tools play a crucial role in computational chemistry and biology, enabling the study of ligand-protein interactions in diverse biological systems.
Docking tools
Algorithms
Benefits
Drawbacks
Reference
Glide
Hierarchical method
Its ability to efficiently explore ligand binding conformations based on geometric complementarity, offering a balance between speed and accuracy in molecular docking.
Its limited ability to account for dynamic flexibility in ligands and receptors, potentially leading to inaccuracies in predicting binding modes in highly flexible systems.
Efficiently explore conformational space, ensuring comprehensive coverage and enhancing the likelihood of identifying precise ligand binding poses.
It may struggle to efficiently sample highly flexible ligands or receptor conformations, potentially leading to missed binding modes or inaccurate predictions in systems with significant conformational variability.
Docking rigid body proteins predicts how proteins and ligands interact, determining their energy. Worldwide, researchers explore diverse protein-ligand systems to understand their interactions better.
It offers a constrained perspective on frequencies in terms of signal processing.
Utilize the advantages of parallelization for both rigid and flexible ligand applications.
For time-dependent methods, such as those used in molecular dynamics simulations, it is not appropriate. There is a problem with the GA’s convergence uncertainty.
Docking tools, algorithms, benefits, and drawbacks.
4.1 GLIDE
Glide is a widely used molecular docking software developed by Schrödinger. It is renowned for its accuracy in predicting ligand-protein interactions and its efficient performance in virtual screening studies. It offers two precision modes: SP (Standard Precision) and XP (Extra Precision). These modes provide varying levels of accuracy and computational efficiency, allowing users to tailor their docking studies to their specific needs. Glide exploits advanced SF to evaluate and rank ligand binding poses. These SF consider various factors such as steric clashes, hydrogen bonding, and van der Waals interactions to assess the quality of ligand-protein interactions and can handle flexible ligands and protein receptor structures, allowing for more realistic modeling of molecular interactions. Glide is capable of efficiently screening large libraries of compounds to identify potential drug candidates or lead molecules [57]. Schrödinger provides a user-friendly graphical interface for setting up and running docking experiments with Glide, making it accessible to both experts and beginners in computational drug discovery.
4.2 AutoDock Vina
It is a powerful tool for predicting ligand-receptor interactions in drug discovery. Dr. Oleg Trott developed AutoDock Vina at The Scripps Research Institute. It estimates binding affinities between ligands and target proteins. The most recent stable version is 1.1.21. AutoDock Vina integrates GA and empirical scoring to efficiently explore ligand conformational space, considering steric clashes and electrostatic interactions for binding affinity estimation. Unlike rigid methods, it permits ligand partial flexibility, enhancing prediction accuracy. Moreover, it identifies potential binding sites in target proteins, aiding researchers in focusing on relevant regions. Recent advancements include QuickVina2 with enhanced search algorithms, Smina offering a user-friendly interface and customizable scoring terms, Vina-Carb optimized for carbohydrate docking, VinaXB addressing halogen bonding, and Vinardo incorporating an improved SF.
4.3 FlexX
FlexX is another molecular docking software, primarily used for ligand docking studies. Developed by BioSolveIT, FlexX offers several features tailored for docking small molecules into protein binding sites, particularly its ability to handle flexible ligands and protein structures. It can account for conformational changes in both the ligand and the receptor during docking simulations, which is crucial for accurately predicting ligand binding modes. FlexX employs SF to evaluate and rank ligand poses within the protein binding site. These SFs consider various factors, such as steric clashes, hydrogen bonding, and electrostatic interactions, to assess the quality of ligand-protein interactions. FlexX is designed to rapidly explore the conformational space of ligands and protein receptors, making it suitable for high-throughput virtual screening studies. FlexX provides a user-friendly interface for setting up docking experiments and analyzing results. It offers visualization tools to help users understand and interpret docking outcomes. FlexX allows users to customize various parameters of the docking process, such as search algorithms, SF, and constraints, to tailor the docking studies to their specific research needs [58].
4.4 GOLD
GOLD (Genetic Optimization for Ligand Docking) is a molecular docking software developed by the CCDC (Cambridge Crystallographic Data Centre). It is widely used in the pharmaceutical industry and academic research for virtual screening, lead optimization, and structure-based drug design. GOLD employs a GA to explore ligand conformational space and optimize ligand binding poses within protein binding sites. The GA mimics natural selection processes to iteratively improve ligand binding affinity and geometry. In molecular docking, GOLD uses SF to rank ligand poses based on their predicted binding affinity and compatibility with the protein’s binding site. These SFs take into account factors like steric clashes, hydrogen bonding, and hydrophobic interactions. It can handle flexible ligands and protein receptor structures, allowing for accurate modeling of conformational changes during ligand binding. It can also incorporate flexibility into specific regions of the protein binding site to improve docking accuracy. The latest iteration of GOLD Suite 5.2 encompasses three key components: Gold 5.2 facilitates protein-ligand docking, Hermes 1.6 enables comprehensive protein visualization, and Gold Mine 1.5 offers tools for the analysis of docking results, including grading.
4.5 LeDOCK
LeDock is a molecular docking software developed by the Laboratory of Structural Bioinformatics at the University of Paris Diderot and the Molecular Modeling Group (CNRS, France). LeDock employs a combination of geometric matching and optimization algorithms for molecular docking. It utilizes geometric complementarity to match ligand conformations to the protein binding site and optimizes ligand poses to improve binding affinity. Evaluation and ranking of ligand poses are carried out by LeDock using SF, considering factors such as steric clashes, hydrogen bonding, and electrostatic interactions. It accommodates flexible ligands and protein receptor structures, allowing for accurate modeling of conformational changes during ligand binding. Robust performance has been demonstrated in virtual screening and lead optimization studies across various protein targets. It has been validated against experimental data and benchmarked against other docking software. LeDock is open-source software, that grants users access to and modification of the source code for customization or integration into larger computational pipelines [59].
An SF is a computational model used in molecular docking to evaluate and rank the binding interactions between a ligand and a protein. The SF takes into account various molecular characteristics, such as the steric fit, electrostatic interactions, hydrogen bonding, and other factors that contribute to the stability of the ligand-protein complex. The primary goal of an SF is to predict and prioritize the binding affinity or strength of different ligands to a given target, helping researchers identify potential drug candidates or understand the molecular basis of biological processes. Different molecular docking programs may employ distinct SFs, and their accuracy is crucial for the reliability of the virtual screening results [60].
5.1 Physics-based scoring functions
A physics-based SF is a sophisticated framework that analyses molecular interactions with a high degree of accuracy. It comprises three integral components, each contributing to a comprehensive understanding of ligand-protein complexes: force field, solvent model, and quantum mechanics. The force field aspect employs a mathematical model to calculate the potential energy of a molecular system. It includes parameters for bond stretching, angle bending, and dihedral angle rotation, providing a classical representation of molecular mechanics. By capturing the geometric aspects and internal energy of the ligand-protein complex, the force field enhances the overall accuracy of the SF. Solvent models are incorporated to account for the impact of the surrounding environment, typically water, on the molecular system. These models consider the effects of solvation on the complex’s energetics. Integrating solvent models into the SF enhances the realism of the molecular environment, taking into account the impact of water molecules on the stability of complexes. This incorporation acknowledges the role of solvent effects in influencing the interactions within the molecular system. Quantum mechanics methods, such as DFT (density functional theory) or ab initio calculations, are employed to compute the electronic structure of the ligand-protein complex. The quantum mechanics component provides a highly accurate representation of electronic interactions, including bond formation, charge distribution, and electronic energy contributions, contributing to a detailed assessment of molecular energetics [61].
5.2 Empirical scoring functions
Empirical SF evaluates a complex’s binding affinity by combining important energetic components associated with protein-ligand binding, such as steric clashes, hydrophobic effects, and hydrogen bonds, amongst others. Usually, linear regression analysis is employed to enhance the weights of these energy components using a training set with known binding affinities. Empirical SFs encompass two primary research directions. The first direction involves the utilization of extensive and high-quality training datasets to enhance the optimization of protein-ligand structures. The second direction focuses on selecting suitable energy terms through stepwise variables and systematic considerations for the target protein. Whilst empirical SFs break down protein-ligand binding affinities into distinct energy terms, akin to physics-based SFs, they typically adopt a flexible and intuitive functional form rather than relying on the well-established models employed by physics-based SFs. Due to their straightforward energy terms, these SFs excel in predicting binding affinity, ligand pose, and virtual screening with minimal computational expense. However, they are less adept at elucidating the correlation between binding affinity and crystal structures, and they often face challenges related to double-counting issues [62].
5.3 Knowledge-based scoring functions
Knowledge-based SFs are computational models used in molecular docking and structure-based drug design to predict the binding affinity of a ligand to a target protein. Unlike physics-based SFs that rely on theoretical models of molecular interactions, knowledge-based SFs derive their information from experimental data and existing knowledge about protein-ligand interactions. The key idea behind knowledge-based SFs is to utilize statistical analysis of experimental structures to identify patterns and preferences in protein-ligand interactions. These functions are trained on databases of experimentally determined protein-ligand complexes, such as the PDB (Protein Data Bank). The statistical analysis involves extracting information about the frequency and distribution of various molecular interactions observed in these complexes [63].
This is because SFs share several common features. Firstly, they predominantly focus on pairwise interactions between specific atoms or atom types in both the ligand and the protein. This allows the SF to pinpoint and evaluate the individual contributions of these interactions. Secondly, these SFs rely on statistical potentials to assess the likelihood of observing a particular interaction at a given distance or angle. By drawing on statistical analyses of experimental structures, they derive insights into the preferences and tendencies of real protein-ligand interactions. Additionally, knowledge-based SF takes into account the distributions of distances and angles between interacting atoms observed in experimental structures. This consideration of spatial arrangements contributes to a more accurate assessment of the likelihood and strength of various interactions. Furthermore, the SF encompasses energy terms that encompass a range of molecular forces and interactions. These terms may include contributions from van der Waals interactions, hydrogen bonding, electrostatic interactions, and other relevant energy components, providing a comprehensive evaluation of the molecular binding. Lastly, some knowledge-based SF incorporates a reference state, which represents the expected behavior of non-interacting atoms. This reference state serves as a baseline for evaluating the significance and favourability of observed interactions within the context of the specific molecular environment [64].
5.4 Machine learning-based scoring functions
Machine learning-based SF is a computational model that utilizes machine learning techniques to predict the binding affinity or likelihood of interaction between a protein and a ligand in molecular docking studies. Molecular docking is a computational method used in drug discovery to analyze and predict the preferred orientation and conformation of a ligand when binding to a target protein. These models are trained on datasets containing information about known protein-ligand complexes as shown in Figure 2, including their experimentally measured binding affinities. The machine learning algorithm learns patterns and relationships between various features or descriptors associated with these complexes. These features could include structural, energetic, or physicochemical properties of the protein and ligand. Once trained, the SF can be applied to predict the binding affinity of new protein-ligand interactions that were not part of the training dataset. The goal is to provide accurate and efficient predictions, aiding in the identification of potential drug candidates or understanding the strength of interactions between biological molecules. Common machine learning algorithms used for developing SF include linear regression, support vector machines, random forests, and more recently, deep learning approaches such as neural networks. Machine learning-based SFs are valuable tools in drug discovery and computational biology, helping researchers prioritize and analyze potential drug candidates more effectively [65].
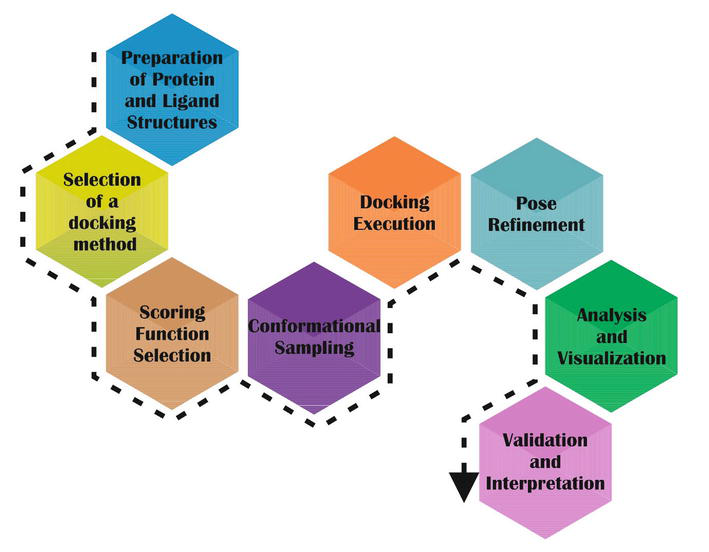
Outlined below are the key stages in molecular docking, a pivotal process in structure-based drug design. These steps systematically guide the preparation, execution, and analysis of ligand-protein interactions, providing a roadmap for elucidating molecular binding mechanisms for a visual representation of these steps, refer to Figure 3, which illustrates the general workflow of molecular docking:
Preparation of protein and ligand structures: Retrieve or generate the 3D structures of the protein receptor and the ligand molecule to be docked. Prepare the protein structure by removing water molecules, heteroatoms, and co-crystallized ligands. Also, check and correct any structural irregularities. Prepare the ligand structure by adding hydrogen atoms, assigning proper charges, and optimizing its geometry [66, 67].
Selection of a docking method: It hinges on the researcher’s requirements. For instance, if docking numerous molecules at a protein’s binding site under specific conditions like pH, water presence, and solubility is the aim, flexible docking programs might be favored. However, when scanning thousands of compounds from databases, flexible docking methods could be impractical unless there is ample processing power and a speedy computer. Hence, users can opt for various docking methods based on their computer’s capabilities and the properties of the target [68].
Scoring function selection: Choose an appropriate SF or scoring scheme to evaluate the fitness of ligand poses within the binding site. SF assesses the binding affinity based on various parameters such as van der Waals interactions, electrostatics, hydrogen bonding, and solvation effects.
Conformational sampling: Perform a systematic search of the ligand conformational space to explore different possible binding poses within the defined search space. Use algorithms such as GA, MC methods, or molecular dynamics simulations to generate and optimize ligand conformations [69].
Docking execution: Apply a docking algorithm to position the ligand within the binding site based on the calculated energy scores. Commonly used docking algorithms include AutoDock, DOCK, Vina, Glide, and GOLD, amongst others [70].
Pose refinement: Refine the initial ligand poses obtained from docking algorithms to improve accuracy and reliability. Use methods like molecular dynamics simulations, energy minimization, or rescoring techniques to refine the ligand-protein interactions [71].
Analysis and visualization: Analyze the docked poses to identify potential binding modes and interactions between the ligand and the protein receptor. Visualize the docked complexes using molecular visualization software to interpret the results and make informed decisions regarding ligand design or further experimental validation [72].
Validation and interpretation: Validate the docking results by comparing them to experimental data, if available. Interpret the binding poses and interactions to understand the molecular mechanisms underlying ligand binding and to guide further experimental studies or drug design efforts [73].
In summary, this chapter provides a thorough exploration of molecular docking software in drug discovery within the realm of computational biology and drug design. Molecular docking emerges as a vital tool, predicting interactions between small molecules and target proteins. Over the last three decades, its evolution has been propelled by computational advancements and increased database accessibility. Our discussion highlights molecular docking’s pivotal role in drug development, from structure–activity studies to lead optimization and virtual screening. These software tools, including AutoDock, GOLD, and DOCK, play crucial roles in understanding molecular interactions. We explore algorithms and search strategies, ranging from systematic approaches such as conformational search to stochastic methods like MC and GA. Shape matching algorithms, MC simulations, particle swarm optimization, and GA are investigated, providing insights into their applications and benefits. SF, spanning physics-based, empirical, knowledge-based, and machine learning-based approaches are highlighted for their roles in evaluating binding interactions. This chapter serves as a guide for researchers, empowering them to navigate the complexities of molecular docking for innovative drug discoveries in the ever-evolving field of computational biology.
References
1.Hu L, Benson ML, Smith RD, Lerner MG, Carlson HA. Binding MOAD (Mother of All Databases). Proteins: Structure, Function and Genetics. 2005;60(3):333-340. DOI: 10.1002/PROT.20512
2.Ahmed A, Mam B, Sowdhamini R. DEELIG: A deep learning approach to predict protein-ligand binding affinity. Bioinformatics and Biology Insights. 2021;15:2-5. DOI: 10.1177/11779322211030364
3.Roche O, Kiyama R, Brooks CL. Ligand-protein database: Linking protein-ligand complex structures to binding data. Journal of Medicinal Chemistry. 2001;44(22):3592-3598. DOI: 10.1021/JM000467K/ASSET/IMAGES/LARGE/JM000467KN00001.JPEG
4.Cournia Z, Allen B, Sherman W. Relative binding free energy calculations in drug discovery: Recent advances and practical considerations. Journal of Chemical Information and Modeling. 2017;57(12):2911-2937. DOI: 10.1021/ACS.JCIM.7B00564
5.Maruca A et al. Computer-based techniques for lead identification and optimization i: Basics. Physical Sciences Reviews. Jun 2019;4(6):4-7. DOI: 10.1515/PSR-2018-0113
6.Kontoyianni M. Docking and virtual screening in drug discovery. Methods in Molecular Biology. 2017;1647:255-266. DOI: 10.1007/978-1-4939-7201-2_18
7.Hollingsworth SA, Dror RO. Molecular dynamics simulation for all. Neuron. 2018;99(6):1129-1143. DOI: 10.1016/j.neuron.2018.08.011
8.Pozzan A. Molecular descriptors and methods for ligand based virtual high throughput screening in drug discovery. Current Pharmaceutical Design. 2006;12(17):2099-2110. DOI: 10.2174/138161206777585247
9.Hawkins PCD, Skillman AG, Nicholls A. Comparison of shape-matching and docking as virtual screening tools. Journal of Medicinal Chemistry. 2007;50(1):74-82. DOI: 10.1021/JM0603365
10.Kuntz ID, Blaney JM, Oatley SJ, Langridge R, Ferrin TE. A geometric approach to macromolecule-ligand interactions. Journal of Molecular Biology. 1982;161(2):269-288. DOI: 10.1016/0022-2836(82)90153-X
11.Younus S, Vinod Chandra SS, Nair ASS. Docking and dynamic simulation study of Crizotinib and Temozolomide drug with Glioblastoma and NSCLC target to identify better efficacy of the drug. Future Journal of Pharmaceutical Sciences. Dec 2021;7(1):6-8. DOI: 10.1186/S43094-021-00323-2
12.Jain AN, Nicholls A. Recommendations for evaluation of computational methods. Journal of Computer-Aided Molecular Design. 2008;22(3-4):133-139. DOI: 10.1007/S10822-008-9196-5
13.Kolb P, Irwin J. Docking screens: Right for the right reasons? Current Topics in Medicinal Chemistry. 2009;9(9):755-770. DOI: 10.2174/156802609789207091
14.Kuan J, Radaeva M, Avenido A, Cherkasov A, Gentile F. Keeping pace with the explosive growth of chemical libraries with structure-based virtual screening. Wiley Interdisciplinary Review in Computer Molecular Science. Nov 2023;13(6):4-10. DOI: 10.1002/WCMS.1678
15.Freidel MR, Armen RS. Mapping major SARS-CoV-2 drug targets and assessment of druggability using computational fragment screening: Identification of an allosteric small-molecule binding site on the Nsp13 helicase. PLoS One. Feb 2021;16(2):2-5. DOI: 10.1371/JOURNAL.PONE.0246181
16.Arakaki AK, Zhang Y, Skolnick J. Large-scale assessment of the utility of low-resolution protein structures for biochemical function assignment. Bioinformatics. 2004;20(7):1087-1096. DOI: 10.1093/BIOINFORMATICS/BTH044
17.Ferrè F, Ausiello G, Zanzoni A, Helmer-Citterich M. SURFACE: A database of protein surface regions for functional annotation. Nucleic Acids Research. 2004;32:2-3. DOI: 10.1093/nar/gkh054
18.Chen R, Li L, Weng Z. ZDOCK: An initial-stage protein-docking algorithm. Proteins: Structure, Function and Genetics. 2003;52(1):80-87. DOI: 10.1002/PROT.10389
19.Pierce B, Tong W, Weng Z. M-ZDOCK: A grid-based approach for Cn symmetric multimer docking. Bioinformatics. 2005;21(8):1472-1478. DOI: 10.1093/bioinformatics/bti229
20.Sauton N, Lagorce D, Villoutreix BO, Miteva MA. MS-DOCK: Accurate multiple conformation generator and rigid docking protocol for multi-step virtual ligand screening. BMC Bioinformatics. 2008;9(1):1-12. DOI: 10.1186/1471-2105-9-184/FIGURES/3
21.Jain AN. Surflex: Fully automatic flexible molecular docking using a molecular similarity-based search engine. Journal of Medicinal Chemistry. 2003;46(4):499-511. DOI: 10.1021/JM020406H/ASSET/IMAGES/MEDIUM/JM020406HN00001.GIF
22.Temml V, Kutil Z. Structure-based molecular modeling in SAR analysis and lead optimization. Computational and Structural Biotechnology Journal. 2021;19:1431-1444. DOI: 10.1016/j.csbj.2021.02.018
23.Bitencourt-Ferreira G, Pintro VO, de Azevedo WF. Docking with AutoDock4. Methods in Molecular Biology. 2019;2053:125-148. DOI: 10.1007/978-1-4939-9752-7_9
24.Fu Y, Wu X, Chen Z, Sun J, Zhao J, Xu W. A new approach for flexible molecular docking based on swarm intelligence. Mathematical Problems in Engineering. 2015;34:2-6. DOI: 10.1155/2015/540186
25.Gardiner EJ, Willett P, Artymiuk PJ. Protein docking using a genetic algorithm. Proteins: Structure, Function and Genetics. 2001;44(1):44-56. DOI: 10.1002/PROT.1070
26.Liu M, Wang S. MCDOCK: A Monte Carlo simulation approach to the molecular docking problem. Journal of Computer-Aided Molecular Design. 1999;13(5):435-451. DOI: 10.1023/A:1008005918983
27.Kodchakorn K, Poovorawan Y, Suwannakarn K, Kongtawelert P. Molecular modelling investigation for drugs and nutraceuticals against protease of SARS-CoV-2. Journal of Molecular Graphics & Modelling. Dec 2020;101:2-6. DOI: 10.1016/j.jmgm.2020.107717
28.Shoichet BK, Kuntz ID, Bodian DL. Molecular docking using shape descriptors. Journal of Computational Chemistry. 1992;13(3):380-397. DOI: 10.1002/JCC.540130311
29.Yamagishi M, Martins N, Neshich G, et al. A fast surface-matching procedure for protein–ligand docking. Springer. 2006;12(6):965-972. DOI: 10.1007/s00894-006-0109-z
30.Van Der Spoel D, Lindahl E, Hess B, Groenhof G, Mark AE, Berendsen HJC. GROMACS: Fast, flexible, and free. Journal of Computational Chemistry. 2005;26(16):1701-1718. DOI: 10.1002/JCC.20291
31.Meyer B, Möller H. Conformation of glycopeptides and glycoproteins. Topics in Current Chemistry. 2006;267:187-251. DOI: 10.1007/128_2006_078
32.Jorgensen WL. Efficient drug lead discovery and optimization. Accounts of Chemical Research. Jun 2009;42(6):724. DOI: 10.1021/AR800236T
33.Hart T et al. A multiple-start Monte Carlo docking method. Wiley Online Library. 1992. [Online]. Available from: https://onlinelibrary.wiley.com/doi/abs/10.1002/prot.340130304 [Accessed: January 06, 2024]
34.Read R, Hart T, et al. Monte Carlo algorithms for docking to proteins. Taylor & Francis; [Online] 1994. pp. 3-4. Available from: https://www.tandfonline.com/doi/abs/10.1080/10610279508032529 [Accessed: January 06, 2024]
35.Tüzün B, Saripinar E. Molecular docking and 4D-QSAR model of methanone derivatives by electron conformational-genetic algorithm method. Journal of the Iranian Chemical Society. 2020;17(5):985-1000. DOI: 10.1007/S13738-019-01835-8
36.Katoch S, Chauhan SS, Kumar V. A review on genetic algorithm: Past, present, and future. Multimedia Tools and Applications. 2021;80(5):8091-8126. DOI: 10.1007/S11042-020-10139-6
37.Prasad KK, Ghosh A. A genetic algorithm approach to optimal Asset allocation of defined contribution pension funds: Evidence from India’s national pension system. Compensation and Benefits Review. 2023;35:2-3. DOI: 10.1177/08863687231195498
38.Chen T, Shu X, Zhou H, Beckford FA, Misir M. Algorithm selection for protein–ligand docking: Strategies and analysis on ACE. Scientific Reports 2023. 2023;13(1):1-15. DOI: 10.1038/s41598-023-35132-5
39.McGann M. FRED and HYBRID docking performance on standardized datasets. Journal of Computer-Aided Molecular Design. 2012;26(8):897-906. DOI: 10.1007/s10822-012-9584-8
40.Pagadala NS, Syed K, Tuszynski J. Software for molecular docking: A review. Biophysical Reviews. 2017;9(2):91. DOI: 10.1007/S12551-016-0247-1
41.Agrawal P, Singh H, Srivastava HK, Singh S, Kishore G, Raghava GPS. Benchmarking of different molecular docking methods for protein-peptide docking. BMC Bioinformatics. 2019;19:7-8. DOI: 10.1186/S12859-018-2449-Y
42.Schneidman-Duhovny D, Inbar Y, Nussinov R, Wolfson HJ. PatchDock and SymmDock: Servers for rigid and symmetric docking. Nucleic Acids Research. 2005;33(Web Server issue): W363. DOI: 10.1093/NAR/GKI481
43.Eisen MB, Wiley DC, Karplus M, Hubbard RE. HOOK: A program for finding novel molecular architectures that satisfy the chemical and steric requirements of a macromolecule binding site. Proteins. 1994;19(3):199-221. DOI: 10.1002/PROT.340190305
44.Park H, Lee J, Lee S. Critical assessment of the automated AutoDock as a new docking tool for virtual screening. Proteins: Structure, Function and Genetics. 2006;65(3):549-554. DOI: 10.1002/PROT.21183
45.Thomsen R, Christensen MH. MolDock: A new technique for high-accuracy molecular docking. Journal of Medicinal Chemistry. 2006;49(11):3315-3321. DOI: 10.1021/JM051197E/SUPPL_FILE/JM051197ESI20060314_081922.PDF
46.Gumede NJ. Pathfinder-driven chemical space exploration and multiparameter optimization in tandem with Glide/IFD and QSAR-based active learning approach to prioritize design ideas for FEP+ calculations of SARS-CoV-2 PLpro inhibitors. Molecules. 2022;27(23):8569. DOI: 10.3390/MOLECULES27238569/S1
47.Eberhardt J, Santos-Martins D, Tillack AF, Forli S. AutoDock Vina 1.2.0: New docking methods, expanded force field, and Python bindings. Journal of Chemical Information and Modeling. 2021;61(8):3891-3898. DOI: 10.1021/ACS.JCIM.1C00203
48.Zsoldos Z, Reid D, Simon A, Sadjad SB, Johnson AP. eHiTS: A new fast, exhaustive flexible ligand docking system. Journal of Molecular Graphics & Modelling. 2007;26(1):198-212. DOI: 10.1016/J.JMGM.2006.06.002
49.Ewing TJA, Makino S, Skillman AG, Kuntz ID. DOCK 4.0: Search strategies for automated molecular docking of flexible molecule databases. Journal of Computer-Aided Molecular Design. 2001;15(5):411-428. DOI: 10.1023/A:1011115820450
50.Fogel GB, Cheung M, Pittman E, Hecht D. Modeling the inhibition of quadruple mutant plasmodium falciparum dihydrofolate reductase by pyrimethamine derivatives. Journal of Computer-Aided Molecular Design. 2008;22(1):29-38. DOI: 10.1007/S10822-007-9152-9
51.Hou T, Wang J, Chen L, Xu X. Automated docking of peptides and proteins by using a genetic algorithm combined with a tabu search. Protein Engineering, Design and Selection. 1999;12(8):639-648. DOI: 10.1093/PROTEIN/12.8.639
52.Janson S, Merkle D, Middendorf M. Molecular docking with multi-objective particle swarm optimization. Applied Soft Computing. 2008;8(1):666-675. DOI: 10.1016/J.ASOC.2007.05.005
53.Kellenberger E, Foata N, et al. Ranking targets in structure-based virtual screening of three-dimensional protein libraries: Methods and problems. ACS Publications. 2008;48(5):1014-1025. DOI: 10.1021/ci800023x
54.Singh S, Bani Baker Q , Singh DB. Molecular docking and molecular dynamics simulation. Bioinformatics: Methods and Applications. Jan 2021;18:291-304. DOI: 10.1016/B978-0-323-89775-4.00014-6
55.Goto J, Kataoka R, Hirayama N. Ph4Dock: Pharmacophore-based protein - ligand docking. Journal of Medicinal Chemistry. 2004;47(27):6804-6811. DOI: 10.1021/JM0493818/ASSET/IMAGES/MEDIUM/JM0493818N00001.GIF
56.Wang et al. GM-DockZn: A geometry matching-based docking algorithm for zinc proteins. Europe PMC. [Online]. Available from: https://europepmc.org/article/med/32369579 [Accessed: January 09, 2024]
57.Friesner RA et al. Glide: A new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. Journal of Medicinal Chemistry. 2004;47(7):1739-1749. DOI: 10.1021/JM0306430/SUPPL_FILE/JM0306430_S.PDF
58.Rarey M, Kramer B, Lengauer T, Klebe G. A fast flexible docking method using an incremental construction algorithm. Journal of Molecular Biology. 1996;261(3):470-489. DOI: 10.1006/jmbi.1996.0477
59.Velavan S, Karnan R, NK-A J of Innovative, and undefined 2020, A comparative study on In silico software’s in statistical relation to molecular docking scores. asianjir.com. 1997;3:1-5. Available from: https://asianjir.com/images/issues/BIOINFORMATICS.pdf [Accessed: January 27, 2024]
60.Li J, Fu A, Zhang L. An overview of scoring functions used for protein–ligand interactions in molecular docking. Interdisciplinary Sciences. 2019;11(2):320-328. DOI: 10.1007/S12539-019-00327-W
61.Meli R, Morris GM, Biggin PC. Scoring functions for protein-ligand binding affinity prediction using Structure-based deep learning: A review. Frontiers in Bioinformatics. 2022;2:2-3. DOI: 10.3389/FBINF.2022.885983
62.Guedes IA, Pereira FSS, Dardenne LE. Empirical scoring functions for structure-based virtual screening: Applications, critical aspects, and challenges. Frontiers in Pharmacology. Sep 2018;9(sep):9-11. DOI: 10.3389/FPHAR.2018.01089
63.Dittrich J, Schmidt D, et al. Converging a knowledge-based scoring function. ACS. 2018;59(1):509-521. DOI: 10.1021/acs.jcim.8b00582
64.Pfeffer P et al. DrugScore RNA knowledge-based scoring function to predict RNA−ligand interactions. ACS. 2007;47(5):1868-1876. DOI: 10.1021/ci700134p
65.Fujimoto KJ, Minami S, Yanai T. Machine-learning- and knowledge-based scoring functions incorporating ligand and protein fingerprints. ACS Omega. 2022;7(22):19030-19039. DOI: 10.1021/ACSOMEGA.2C02822
66.Madhavi Sastry G, Adzhigirey M, Day T, Annabhimoju R, Sherman W. Protein and ligand preparation: Parameters, protocols, and influence on virtual screening enrichments. Journal of Computer-Aided Molecular Design. 2013;27(3):221-234. DOI: 10.1007/S10822-013-9644-8
67.Aja PM et al. Prospect into therapeutic potentials of Moringa oleifera phytocompounds against cancer upsurge: de novo synthesis of test compounds, molecular docking, and ADMET studies. Bulletin of the National Research Centre. Dec 2021;45(1). DOI: 10.1186/S42269-021-00554-6
68.Kitchen DB, Decornez H, Furr JR, Bajorath J. Docking and scoring in virtual screening for drug discovery: Methods and applications. Nature Reviews. Drug Discovery. 2004;3(11):935-949. DOI: 10.1038/nrd1549
69.Elokely KM, Doerksen RJ. Docking challenge: Protein sampling and molecular docking performance. Journal of Chemical Information and Modeling. 2013;53(8):1934-1945. DOI: 10.1021/CI400040D
70.Tiwari R, Prakash K, Pradesh U, Professor A, Tiwari S. Unrevealing the complex interplay: Molecular docking: A Comprehensive review on current scenario, upcoming difficulties. ijcr.info. 2024. 10.22159/ijcr.2024v8i1.226
71.Yamashita T, Sakano T, Iqbal Mahamood M, Fujitani H. Molecular dynamics analysis to evaluate docking pose prediction. jstage.jst.go.jp. 2016;13:181-194. DOI: 10.2142/biophysico.13.0_181
72.Agu PC et al. Molecular docking as a tool for the discovery of molecular targets of nutraceuticals in diseases management. Scientific Reports. 2023;13(1):13398. DOI: 10.1038/S41598-023-40160-2
73.Klepeis JL, Lindorff-Larsen K, Dror RO, Shaw DE. Long-timescale molecular dynamics simulations of protein structure and function. Current Opinion in Structural Biology. 2009;19(2):120-127. DOI: 10.1016/j.sbi.2009.03.004
Written By
Mohd Mursal, Mohammad Ahmad, Sahil Hussain and Mohemmed Faraz Khan
Submitted: 29 January 2024Reviewed: 12 February 2024Published: 29 April 2024